
- AI
- A
Nobel Prizes 2024 and Artificial Intelligence. Chemistry: Protein Structure Prediction
With you again is Pavel Buzin from Cloud.ru. As promised in the first part of the Nobel Prize saga, today we will talk about chemistry. I hope the public's interest in the news has not yet faded, because, I confess, even for me, a technically savvy person, it took quite a bit of time to understand the chemical component of this year's laureates' research.
October 8, 2024 can now be considered one of the most important dates in the history of artificial intelligence, because the Nobel Prize in Chemistry was essentially awarded for the application of AI methods. American David Baker and employees of Google's British subsidiary Demis Hassabis and John M. Jumper took the prize for predicting protein structures. Stop. Since when did Google become a chemical company? In this article, we will look at the background of the researchers and the methods they discovered a little more closely.
Demis Hassabis and John Jumper are, respectively, the CEO and director of DeepMind, a Google subsidiary specializing in the development and application of artificial intelligence methods. It is worth mentioning that Nobel Prizes have repeatedly been awarded for discoveries made by employees of corporate research divisions. One can recall Bell Labs (now a division of Nokia Corporation), whose researchers created the first transistor and discovered the cosmic microwave background radiation. Or researchers from IBM who received awards for creating the tunneling microscope and discovering high-temperature superconductivity.
Repeatedly awarded prizes for the development of new tools and methods that expand capabilities and achieve radically new scientific results. A striking example from physics is the Wilson chamber (1927), in chemistry - methods for synthesizing molecules with specified properties "click chemistry" (2022) and much, much more.
Computational methods, which are now collectively referred to as Computer Science, have also been repeatedly recognized with high awards. Most of them are Nobel Prizes in Economics (due to the absence of a mathematics prize), here as an example we can mention Leonid Kantorovich (1975), the father of linear programming, who received the prize with the wording "for his contribution to the theory of optimal resource allocation".
So what did they do this time in the field of computer science that was recognized as revolutionary in chemistry?
Geometric Chemistry
The Nobel Committee's press release states: "The Royal Swedish Academy of Sciences has decided to award the Nobel Prize in Chemistry 2024 ... “for protein structure prediction”.
Already interesting. Protein research is one of the most important areas of modern science, encompassing chemistry, biology, medicine, pharmaceuticals, computer science. In this area, researchers face three major problems:
the complexity and high cost of conducting experiments;
protein molecules consist of long chains made up of amino acids, and the number of their combinations is very large;
like any long molecules, protein molecules have a large number of internal degrees of freedom and, twisting, can take various forms, including those with an internal ordered or disordered structure.
At the same time, different parts of the molecules can geometrically interlock, which is not accompanied by the formation of new chemical bonds. And this already begins to strongly affect the properties of proteins when interacting with other agents — chemical reagents, other proteins, drugs, viruses, various cell elements.
In chemistry, it is not uncommon for the formula of a substance to remain unchanged, but the change in the position of molecules affects how this substance interacts with others. Everyone knows that water and ice are the same H2O molecules, however, ice occupies a larger volume and is less willing to react. Understanding the conditions under which proteins spontaneously change the geometry of their molecules, or knowing what factors initiate this, is extremely important. This affects things like:
shelf life of drugs and reagents;
formation of inactive or even toxic isomers of proteins;
spontaneous change or restoration of the geometry of molecules over time.
The process of turning a chain of amino acids that make up a protein molecule into an ordered structure is called protein folding.
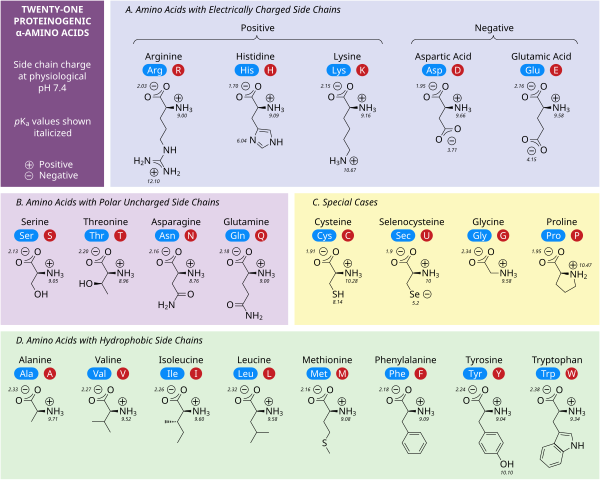
A typical protein molecule is a chain of hundreds and thousands of carbon atoms, having bonds with nitrogen, oxygen, sulfur atoms, cyclic and acyclic compounds, hydroxyl groups, and much more. It was previously impossible to know in advance what spatial origami the amino acid chain would eventually "pack" into. Just imagine: a protein with a chain of 100 amino acids can pack into 1047 different configurations. There are about three dozen amino acids themselves, but the matter is further complicated by the fact that enantiomer amino acids can initially participate in protein synthesis: these are molecules that are mirror images of each other in space (like the right and left hand). Due to their geometric properties, enantiomers interact differently with other molecules. Thus, the total number of amino acid variants, their combinations, enantiomers, and ways to pack all this into a protein molecule exceeds the number of particles in the Universe. Fortunately for researchers, nature is not so diverse, and if you observe the folding and unfolding of amino acid chains "in a test tube," it turns out that they still form a limited number of configurations. The number of options is limited because different spatial configurations of a molecule have different internal energies, and molecules tend to adopt configurations with the lowest internal energy as the most stable.
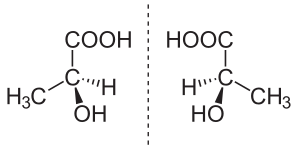
The topic of enantiomers is also important because, in the process of biological evolution on Earth, all proteins adopted only one mirror orientation, called left-handed, which is manifested in the direction of rotation of polarized light when passing through a solution containing an enantiomer. Separation of enantiomers is impossible by physical or chemical means without destroying the molecules. At the same time, mirror copies can be inactive or even toxic, and to obtain a pure enantiomer, it is necessary to use a "seed" in the form of a natural molecule during the synthesis process, which sets the required configuration.
Before diving headfirst into the world of protein chemistry, let's refresh our memory on a few terms that we will need later.
In biochemistry, sequences consisting of amino acids and sugars that do not change during protein synthesis reactions are called residues. The concept of a residue is broader than groups of atoms (such as hydroxyl, carboxyl, or amino groups) because it can have a more complex structure and consist of several groups.
We will also need an understanding of the peptide bond — this is when the amino groups (—NH2) of one amino acid interact with the carboxyl group (—COOH) of another amino acid, establishing a C=N bond with the formation of a free water molecule.
And we need to know about the three main methods of conducting experiments in biology and chemistry: in vivo (in a living organism), in vitro (in a test tube), and in silico (on a computer). Modeling chemical and biological processes is essential due to the complexity and cost of natural experiments.
So, let's go: in silico.
A brief history of proteins "in digits"
One of the largest protein databases — Protein Data Bank (PDP), contains information about 225 thousand proteins and other structures. While the total number of known proteins that are not described in detail is 200 million.
The main methods previously used to predict the shapes of molecules were based on solving equations describing the distribution of electrons in atoms (as a development of approaches to solving the Schrödinger equation) and on the geometric properties of atoms, which can be obtained as experimental data (X-ray, tunneling microscopes, statistical and other methods).
Solving the Schrödinger equation for a single atom made it possible to obtain information about orbitals — single-electron wave functions that provide an idea of the distribution of electrons in an atom. Solving the problem for a two-atom system is still impossible even for the current level of supercomputer development.
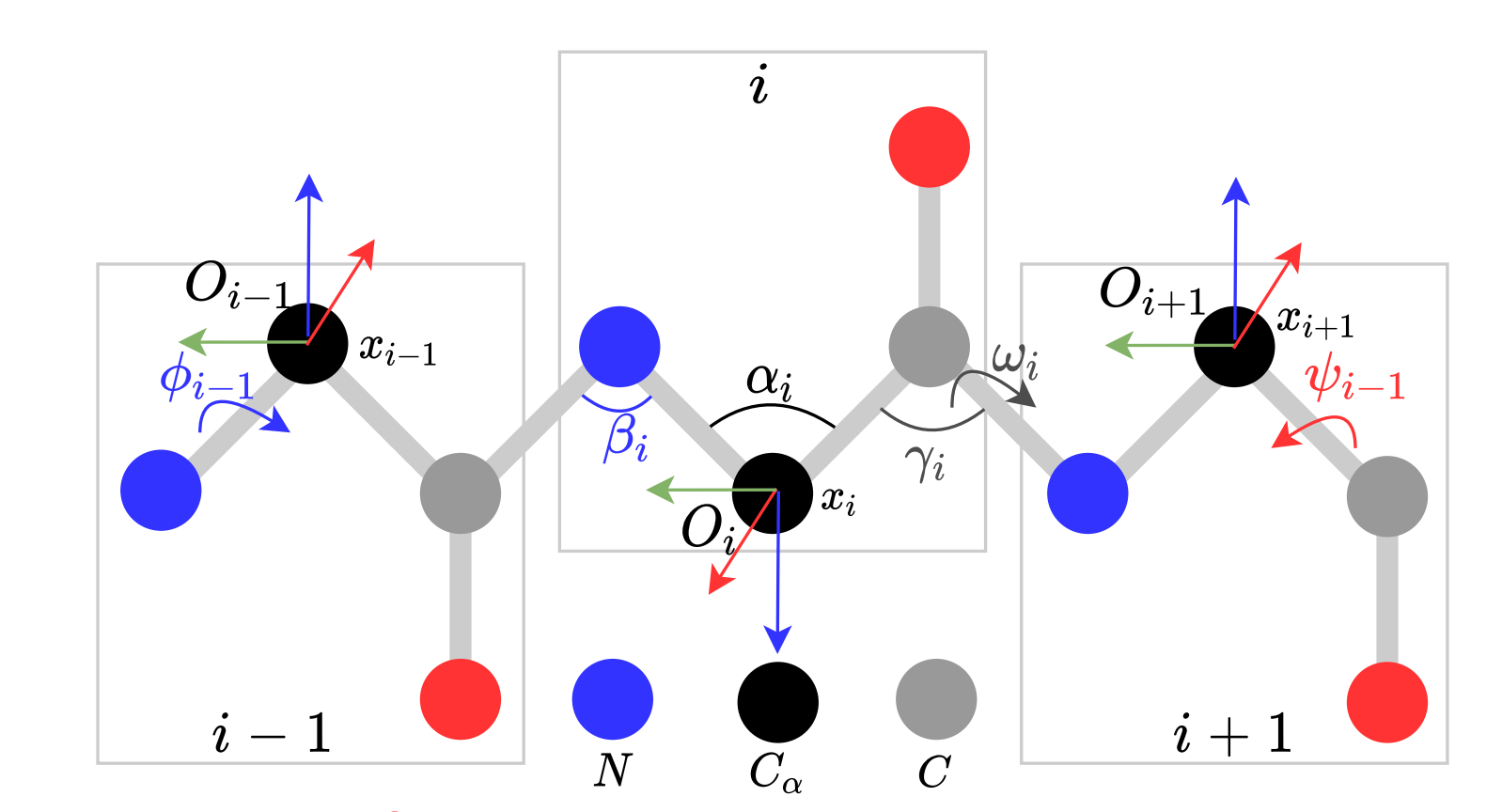
The application of the geometric approach allows us to understand how chemical reactions occur for relatively simple molecules. Due to quantum mechanical effects and temperature fluctuations of atoms in the molecule, the molecule continuously vibrates and changes its shape, taking on some stable states. The figure below shows the possible changes that can occur: changes in the angles between bonds with neighboring atoms (αi, βi, γi, and others), rotation around the interatomic bond (ωi, φi-1, ψi-1), changes in the distance between neighboring atoms (the interatomic bond acts as a spring/oscillator). I note that atoms in the process of movement can take not arbitrary positions, but those that are precisely determined by the energy levels of each molecule.
A small lyrical digression — why are there no protein cyclic or branched structures in nature? As laymen, we do not know (if you know, tell us in the comments). But logically, protein structures in nature are formed in the process of replication. Such processes of spontaneous replication for branched structures are impossible, moreover, they are possible only for a limited fraction of linear and cyclic structures. But in laboratory conditions, branched structures can be synthesized and studied, which is helped by computer modeling.
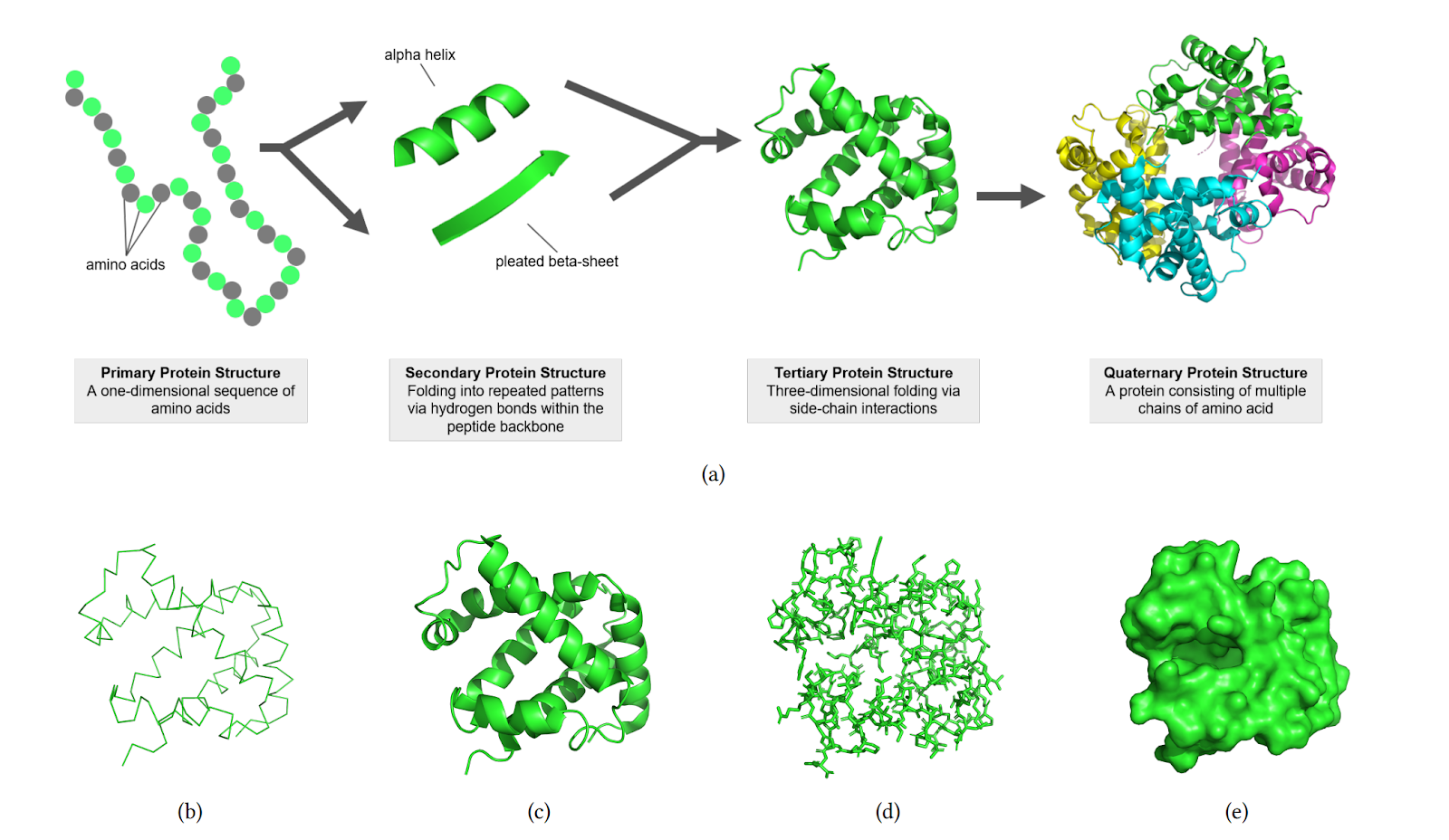
For proteins consisting of a large number of amino acids, an expansion of the abstract description in the form of chemical formulas, to which we are accustomed in chemistry lessons, was required. Biologists and chemists operate with a four-level representation:
Primary Protein Structure — a one-dimensional sequence of amino acids.
Secondary Protein Structure — folding into repeating structures, linear or helical.
Tertiary Protein Structure — three-dimensional folding through the interaction of side atoms and groups of atoms (protruding to the side from the main chain, which consists mainly of carbon atoms).
Quaternary Protein Structure — the protein structure is formed by several chains of interacting amino acids. To visualize the quaternary structure of the protein, representations in the form of lines, ribbons, stick-connections between atoms, and the surface of the electron cloud surrounding the protein molecule are also used.
The figure below illustrates the structures of the human foetal deoxyhaemoglobin protein (PDB: 1FDH)
I think from the above, you are convinced that the study of protein folding is an incredibly complex field. Let's take a short excursion into the history of how protein structure prediction tasks were solved before 2024.
1994 — Critical Assessment of protein Structure Prediction
Today we are already used to hackathons. How about a protein structure folding championship? In 1994, the first CASP championship — Critical Assessment of protein Structure Prediction — was held, and since then it has been held every two years. Teams of researchers receive an amino acid sequence and compete in predicting secondary and tertiary structures for previously unstudied proteins. Neither the organizers, nor the experts, nor the participants know the structure of the test proteins until the prediction stage is completed.
2005—2008 — Rosetta@home and Foldit
Back in 2003, the Human Genome Project sequenced 85% of the human genome. Researchers figured out the amino acid sequences of almost all proteins in the human body and decided: since we have studied everything that exists, let's create in a virtual test tube what does not exist. For example, new more active proteins or ways to change structures responsible for serious diseases.
This is how the Rosetta@home volunteer computing project appeared, where collective computing resources were used by researchers to predict the tertiary structure of proteins and predict the interaction of protein structures. But there was one problem: there are not so many scientists who can come up with new molecules in their free time. And to speed up the victory over cancer and Alzheimer's, enthusiasts decided to popularize protein folding and turn a harsh fun for scientists into a Rubik's cube that everyone can practice with. The main enthusiast of this initiative was this year's future laureate David Baker.
It was he, together with his colleagues, who developed the online puzzle Foldit, where people even without specific knowledge in the field of chemistry can "twist" the amino acid sequence to solve a specific problem. After all, 38,000 heads (the number of Rosetta@home users in 2011) is good, but 240,000 (the number of Foldit players in the year of release) is better. The goal of the puzzle is to find the three-dimensional structure of a specific protein with the lowest level of free energy. Each task is published on the website for a certain period, during which users compete with each other. With the help of Foldit, several scientific breakthroughs have been made: for example, deciphering the structure of the virus that causes AIDS in monkeys, and changing the structure of the protein responsible for catalyzing the Diels-Alder reaction.
2017—2024 — AlphaFold
It's time to talk about the most interesting. Watch closely:
In 2010, a startup called DeepMind Technologies, which focuses on artificial intelligence, was founded in London.
In 2014, the company was acquired by Google.
In 2016, the AlphaZero model, developed by the DeepMind team, won a game of Go against world champion Lee Sedol.
In 2017, AlphaZero achieved the highest chess rating by defeating the strongest chess program at the time, StockFish 8, in a series of 100 games. DeepMind trains a wide family of Alpha models, which achieve brilliant success in various fields.
In 2018, the Alpha family of models was joined by AlphaFold, designed to predict protein structure. The model was developed under the leadership of Demis Hassabis and John Jumper. The research team participated in the 13th CASP competition and took first place.
In 2020, AlphaFold2 once again solved the main CASP task so well that the world's leading scientific journal Nature called it a "breakthrough".
To Google's credit, it has made the AlphaFold and AlphaFold2 models available for use by other researchers, and has also created the AlphaFold Protein Structure Database and filled it with information on 200 million protein structures calculated by DeepMind. For those interested, here are the links to the original sources:
AlphaFold repository: https://github.com/google-deepmind/alphafold
AlphaFold Protein Structure Database: https://alphafold.ebi.ac.uk/
Additional model description: https://pmc.ncbi.nlm.nih.gov/articles/instance/8387230/bin/41586_2021_3819_MOESM1_ESM.pdf
Now about what's inside.
Inside AlphaFold
According to the description, AlphaFold "directly predicts the 3D coordinates of all heavy atoms for a given protein, using the primary amino acid sequence and aligned homologous sequences as input data".
I recommend reading the original sources, you won't regret it
The architecture of AlphaFold2 DeemMind was revealed in an article in the journal Nature, published on July 15, 2021 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8371605/). The most interesting part is in the supplement to this article, in the Supplementary information. Machine learning specialists can immediately follow the link to study it https://pmc.ncbi.nlm.nih.gov/articles/instance/8387230/bin/41586_2021_3819_MOESM1_ESM.pdf.
I will share my opinion on what was important, interesting, really very difficult and breakthrough in AlphaFold2.
The first thing that catches your eye is the training dataset. The DeepMind team used data on 250+ thousand proteins and their properties (such as sizes, configurations, bond angles, distances between atoms, and others) from the Protein Data Bank. In the next step, a separate model was created to generate synthetic examples based on real data (data augmentation) to create the AlphaFold2 training dataset. At the same time, 25% were original examples of proteins from the Protein Data Bank, and 75% were synthetic. When checking the correctness of synthetic examples, the Kullback-Leibler distance was used as a metric for pairs of real example-synthetic example.
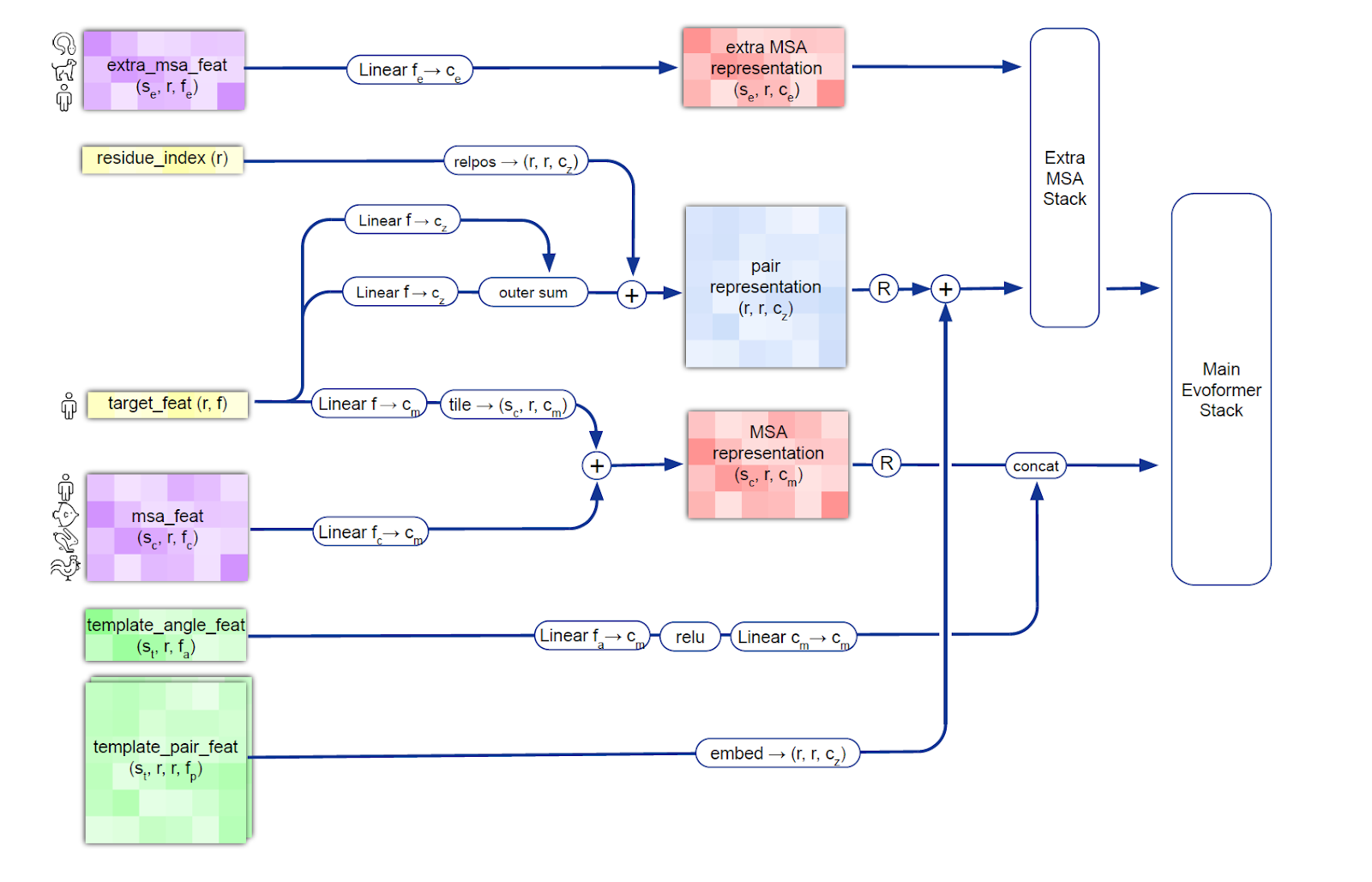
The AlphaFold2 model works with amino acid sequences that make up proteins called multiple sequence alignments (MSA). The AlphaFold2 model receives MSA embeddings, which make up proteins, and features of known pairs of sequences of known proteins (from the training dataset). The network consists of two main modules: the Evoformer module and the structure module.
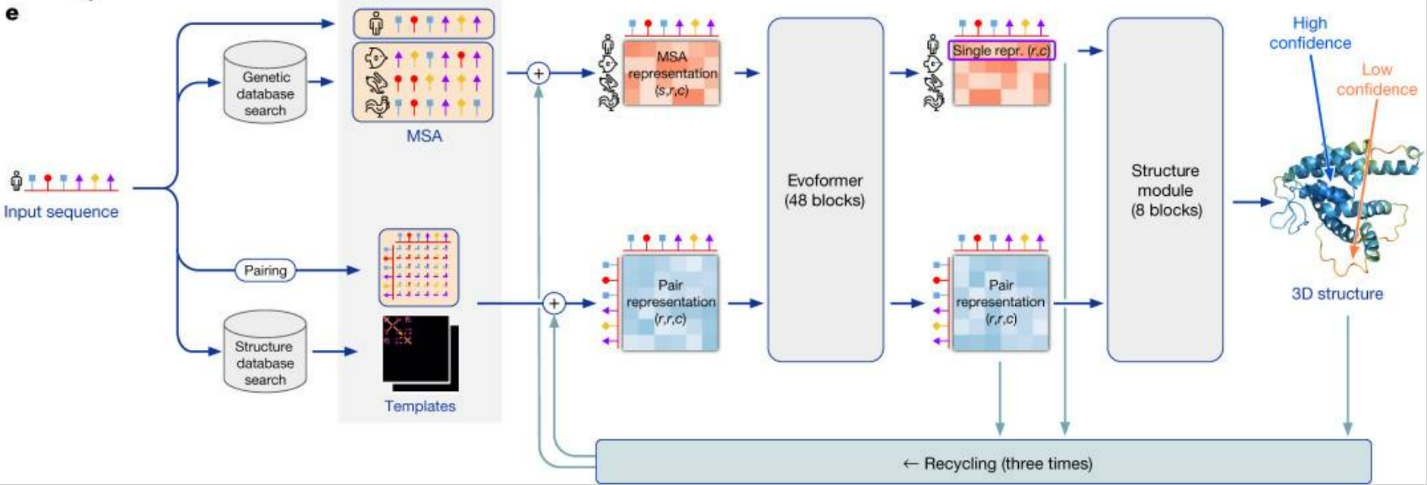
Evoformer operates with MSA embeddings, as well as data on the geometry of real molecules (angles, distances, configuration). The module includes 48 consecutive blocks and uses the attention mechanism, including for calculating angles between atoms. Quoting DeepMind: "the key innovations in the Evoformer block are new mechanisms for exchanging information within MSA and pairwise representations, allowing direct reasoning about spatial and evolutionary relationships." As a result, Evoformer implements a geometric approach to calculating the shape of protein molecules.
As a result, Evoformer outputs:
an Nseq × Nres array, which represents the processed MSA (Nseq is the number of amino acid sequences, Nres is the number of residues - the very groups of atoms we talked about in the "Geometric Chemistry" section);
an Nres × Nres array, which represents pairs of residues.
The Evoformer output is fed into the structure module for reconstruction.
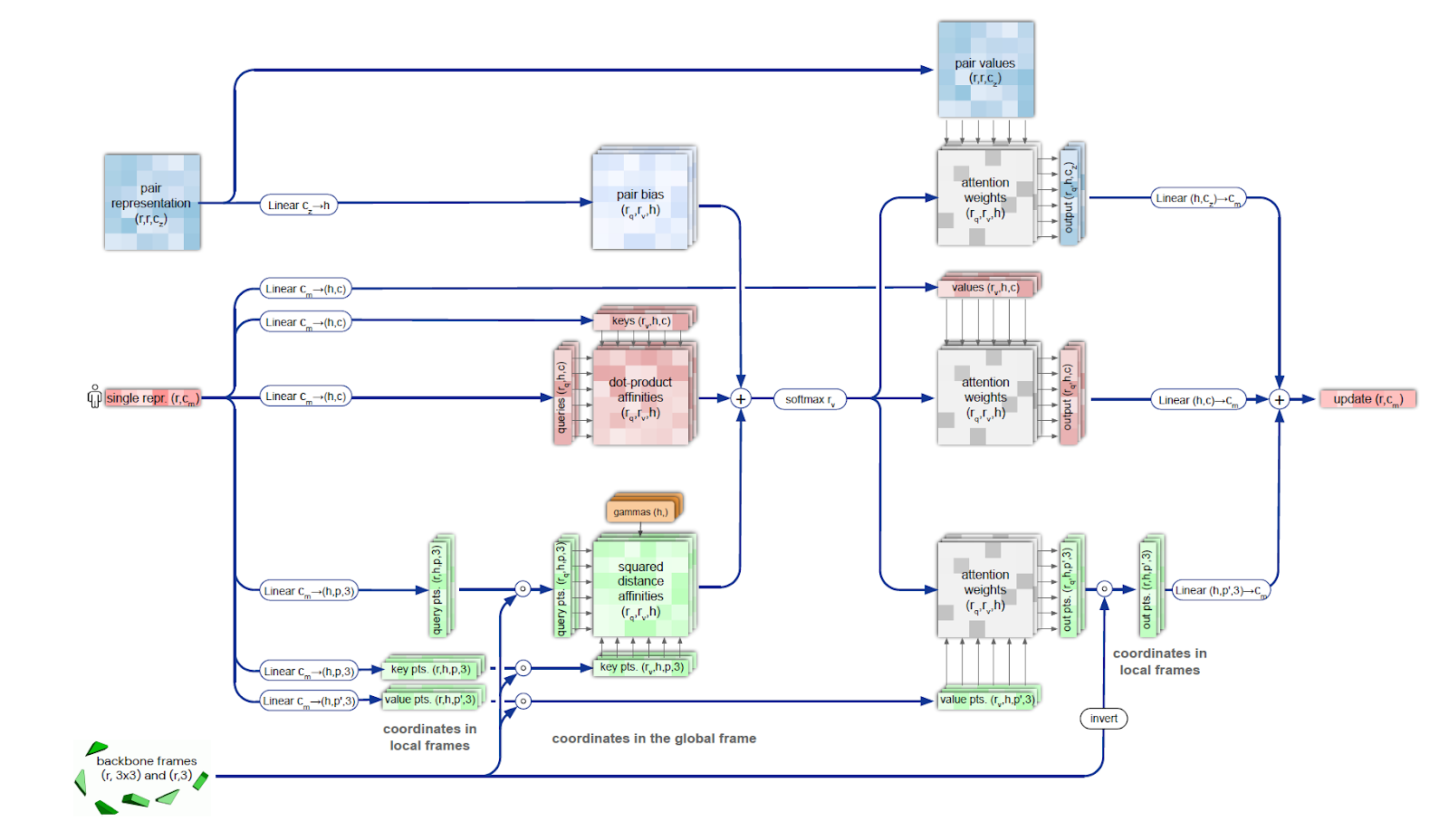
The structure module is used to restore the predicted shape of the molecule by representing the rotation and translation for each protein residue. The module iteratively models the evolution of the initial state (position of atoms and bonds), repeatedly feeding the module's output back into the input. This iterative refinement (the authors call it "recycling") significantly improves accuracy with a slight increase in training time. The Invariant Point Attention (IPA) module ensures the preservation of the N-Cα-C atom sequence in the protein molecule when restoring the molecule's shape.
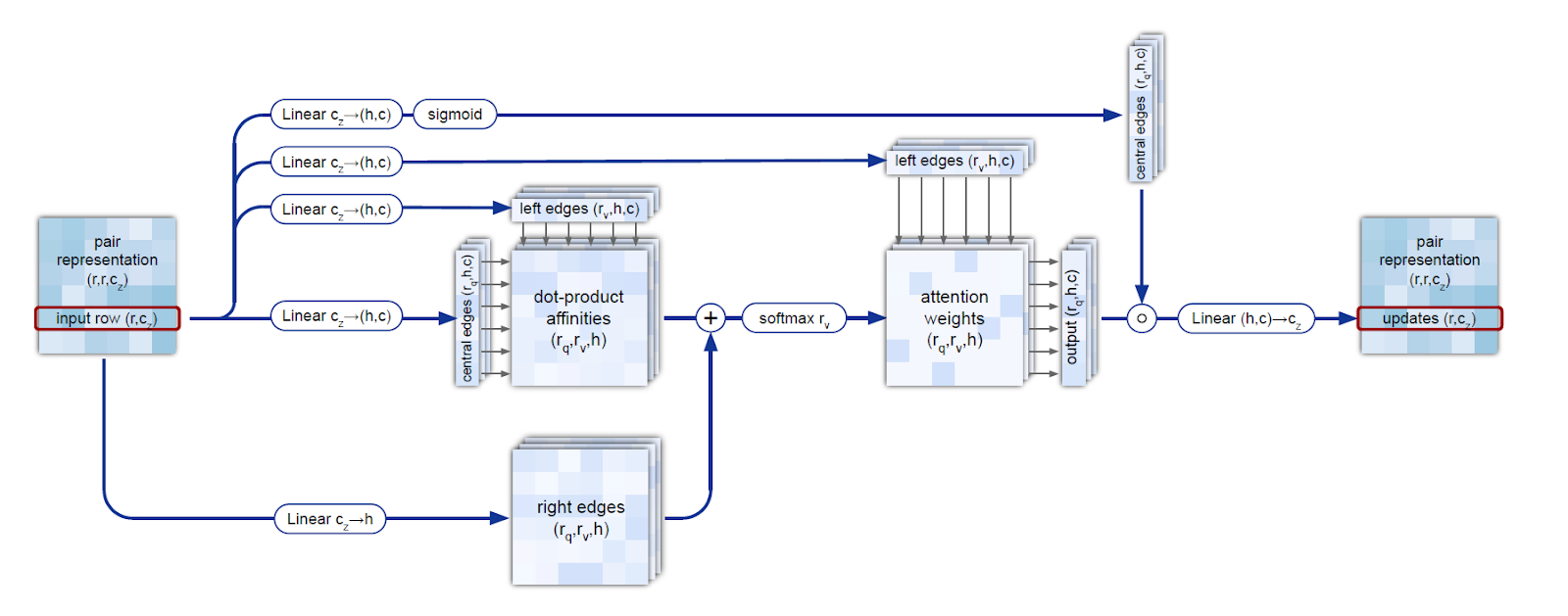
In the process of restoring the 3D structure, many constraints must be met, including the triangle inequality for distances. This is achieved by combining sequential triangle updates and the triangle self-attention module. This process is more accurate and efficient compared to using only the attention mechanism or updating triangles separately.
Quaternions are used in AlphaFold2 to restore the geometry of the predicted protein. This is an extension of complex numbers used in mechanics to describe the motion of a rigid body. Quaternions represent a number in the form q=a+bi+cj+dk, where a, b, c, d are real numbers, and i, j, k are imaginary units with properties i² = j² = k² = ijk = −1.
Quaternions are the pinnacle of higher mathematics. They allow for convenient representation of object rotation in space, simplify calculations, and minimize possible errors in computations. The application of quaternions is a confirmation of the highest level of task elaboration in terms of the geometric properties of molecules.
Also, the structure module implements a mechanism that takes into account the arrangement of neighboring groups of atoms and the peptide bonds arising between them.
As a result, at the output of AlphaFold2, we have data on the position of atoms that make up the protein molecule, the shape of which is modeled by the network.
At the same time, AlphaFold2 achieves very high accuracy and turned out to be much more accurate than competing methods. The median accuracy of AlphaFold2 is 0.96 Å (angstrom, 10-10 meters), which is comparable to the size of a carbon atom 1.4 Å. Moreover, AlphaFold2 can be used to analyze proteins with long chains and domain packing without significant loss of accuracy.
And as the cherry on top, the model is available in open access in the repository with a description of how to deploy the image in Google Cloud. AlphaFold2 is very economical in terms of computational resources - it only requires 12 vCPUs, 85 GB RAM, and one GPU A100 to operate. According to DeepMind, AlphaFold2 has been used more than 2 million times.
Instead of a conclusion
It is often heard that the Nobel Prize was awarded unfairly: sometimes biologists are given the prize for chemistry, sometimes computer scientists for physics. Indeed, David Baker — a bioinformatician and biochemist, has dedicated almost his entire life to protein design and predicting their tertiary structure. The merits of Demis Hassabis and John Jumper lie more in the field of data science, computational biology, and chemistry, and they all received the Nobel Prize for creating neural network-based tools and their applications. Should the public be outraged about this? I would say that we should be happy. The vast amount of cross-disciplinary research and the fact that it is becoming difficult for us to draw a clear line between physics, chemistry, biology, medicine, and computer science speaks more about how deeply we have penetrated the essence of the world around us than about the bias of the Nobel Committee.
The main conclusion that can be drawn from this year's awards: The Nobel Committee and the scientific community as a whole have recorded the emergence of a new reality in which neural networks are just as much a tool in the hands of a researcher as a microscope or a particle accelerator. Previously, science had experimental facts and theories that allowed us to interpret reality and had predictive power. Now the artifacts of science have been supplemented with machine learning models, which are noteworthy, as models are entities that have predictive power but are not interpretable.








Write comment