- AI
- A
FREED++. Accelerating drug discovery with neural networks
Hello! My name is Alexander Telepov, I am a researcher at the AIRI Institute. Our team is engaged in the application of deep learning in life sciences. Our interests include tasks such as material design, solubility analysis, or the search for new drugs. I would like to talk more about the latter.
Many have heard that neural networks are used today to search for new compounds. Take, for example, the sensational AlphaFold 3 from DeepMind, which solves the problem of generating the three-dimensional structure of various molecular complexes. There are other tasks in which neural networks have outperformed classical numerical methods. The most striking example is the generation of drug molecules. One of the most notable approaches to this task was the drug molecule generation framework based on reinforcement learning methods FREED. But it was far from perfect.
Not so long ago, our research group reproduced, thoroughly investigated, and significantly improved FREED. We will present our results in the journal TMLR, the article is available on arXiv. Here I will briefly talk about FREED itself and its problems, as well as the essence of our improvements to this approach.
Drug Development
It is difficult to meet a person today who has never taken any medicine. It is equally difficult to overestimate the importance of pharmacology and the science of drug development. For the sake of clarity, we will call drugs substances used in the prevention, diagnosis, and treatment of diseases.
As a rule, such development follows the "drug-target" scheme in three stages:
-
First, the target protein is identified — a key molecule involved in a specific metabolic or signaling pathway associated with a particular disease condition.
-
The three-dimensional structure of the protein is determined (usually by crystallography methods).
-
A molecule with the desired set of properties that selectively interacts with the target protein is found.
The latter task is the subject of study in the field of medicinal chemistry. In the simplest case, the main task of medicinal chemistry can be formalized as follows:
To propose a chemical compound (a small molecule, ligand) that, when binding to the protein of interest, significantly changes its functional state.
Quantitatively, this effect is expressed by the change in Gibbs energy for the protein-ligand complex
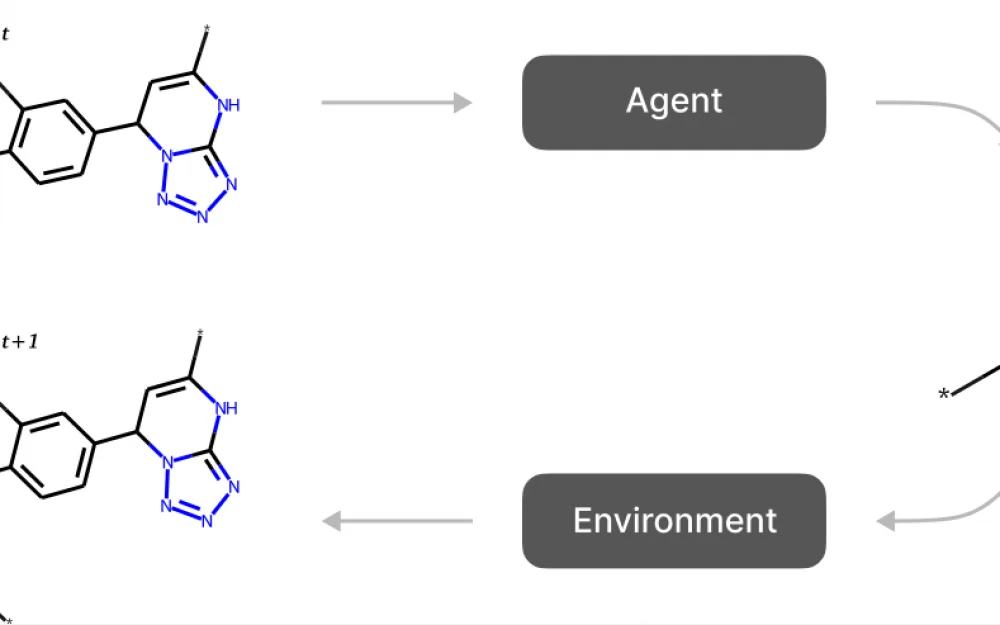
Fig. 1. Scheme of generative methods based on reinforcement learning. The agent takes the current state as input and selects an action. The action usually consists of attaching a molecular fragment to the state assembled so far.
It should be noted that until the FREED model appeared (i.e., the end of 2021), the vast majority of works on molecular generation (CVAE, ORGAN, REINVENT, GraphVAE, MolGAN, JT-VAE, GCPN) considered the task of unconditional generation, one in which the target protein was not taken into account (although there were exceptions — LiGAN). Such models are not optimal, as the therapeutic effect of a molecule is significantly determined by its binding energy with the target protein.
FREED Method
At the end of 2021, a promising approach to drug molecule generation based on reinforcement learning methods, FREED, was published at the NeurIPS conference. Unlike most previous works, FREED takes into account the biological target protein when generating ligands. In addition, the method directly optimizes the binding energy of the protein with the ligand. Let's note a few more useful properties of FREED:
-
Fragment generation significantly reduces the space of possible actions and states, allowing efficient training of the RL agent;
-
FREED works on an extended molecular graph — a graph in which there are special vertices, attachment points (these vertices determine the places where molecular bonds were broken during fragmentation). If fragments are attached only to these points, the assembled molecule will automatically satisfy the valence rules, provided that the missing hydrogens are added to the molecule after the terminal step.
The results described in the FREED article are indeed impressive. However, during the course of conducting a large number of experiments and thoroughly analyzing the publication's source code, we found that the proposed method has significant limitations and problems. Namely:
-
there are multiple bugs in the existing implementation;
-
the comparison protocol is inconsistent between the proposed model and the baselines;
-
the number of selected protein targets for quality assessment is insufficient;
-
the model is overly complex and the effects of its individual parts are underexplored.
From FREED to FFREED and FREED++
To eliminate the limitations of FREED, we thoroughly investigated the publication's source code, fixed the errors, conducted additional ablations, and simplified the proposed model.
Before describing the changes made to the original model, it is necessary to describe it in more detail. However, since the original model contains bugs and inconsistent blocks, this may mislead the reader. Therefore, I will first describe how the corrected FFREED (Fixed FREED) model is arranged, and then give an example of one of the bugs found in the model.
FFREED
Let's consider how the action selection (policy architecture, actor) and the state-action value function (critic) are arranged in FFREED.
An action is a composition of three elements. The first component is the attachment point on the molecule assembled at this step. A new fragment will be attached to this point. The second component is the fragment that will be attached. It is selected from a fixed dictionary of fragments obtained during the fragmentation procedure. And finally, the third component is the attachment point on the fragment.
The action selection process is shown in Fig. 2a.
Step 1: current state
Fig. 2. Schematic architectures of the actor and critic in the FFREED model.
Below we describe one of the most significant bugs related to the parameterization of the critic (state-action value function).
In the original implementation, the critic is parameterized by a multilayer perceptron that takes as input the concatenation of 4 vectors:
1) the embedding of the molecule obtained by the graph neural network;
2) the vector of probabilities associated with the attachment points on the molecule;
3) the one-hot representation of the selected fragment;
4) vectors of probabilities associated with attachment points on the selected fragment.
In this implementation, there are two significant problems. First, the critic does not receive any information about the selected attachment points, as it only operates on the probability distribution over these points. Second, the order in which the probabilities themselves are fed into the critic is determined by the internal representation of the current state. This order can change when new fragments are attached along the trajectory. Thus, the critic cannot associate high rewards with the selection of specific attachment points. Accordingly, the task of learning any reasonable policy for selecting attachment points becomes unsolvable. In our implementation, instead of probability vectors, we feed embeddings of the selected attachment points into the critic.
After fixing all the bugs, the model successfully handles reward optimization, as can be seen in the training graph, see Fig. 3.
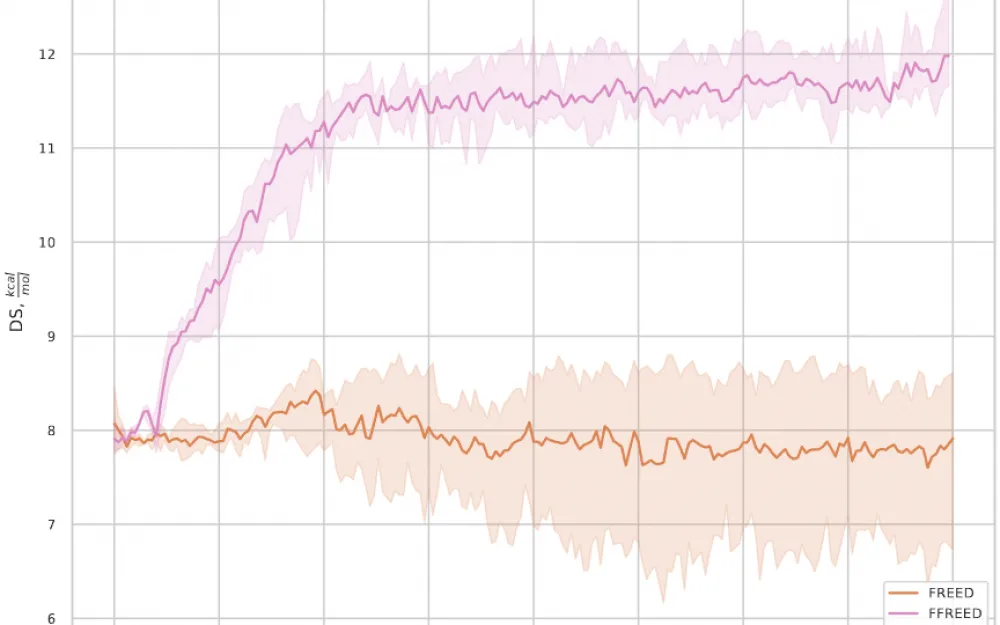
Additionally, in the ablations, we investigated how the elimination of each bug in the original FREED model affects the quality of generation. It turned out that fixing any single bug alone does not lead to a significant improvement in quality — all errors need to be fixed together.
FFREED++
At the next step, we significantly accelerated the FFREED model and reduced the number of trainable parameters in it. We called the resulting model FREED++. Let me explain in order what exactly we did.
Policy Architecture. Let's take a closer look at how the policy architecture is arranged in the FREED and FFREED models. The action selection occurs autoregressively, and each component of the action
Fig. 4. Scheme for calculating the conditional probability of actions for FFREED and FREED++.
Each component of the policy computes the probability distribution over the set of available options under some condition. For example, for
Table 1. Comparison of FFREED and FREED++ speeds. Time denotes the number of seconds per 1 gradient descent step per batch size of 100.
Comparison with Baselines
After fixing errors in the source code and optimizing the model, we compared our result with other existing models on the task of generating molecules with high affinity. In the original work, the comparison protocol between baselines and the FREED model is inconsistent, and the test protein set is insufficient for accurate comparison. In our work, we expanded the test protein set from 3 to 6, performed hyperparameter tuning for the baselines, added 2 methods for comparison (combinatorial generator and Pocket2Mol, which is still considered a strong baseline today), and also increased the computational budget for baseline models (in the original article, the REINVENT and MolDQN models were significantly undertrained).
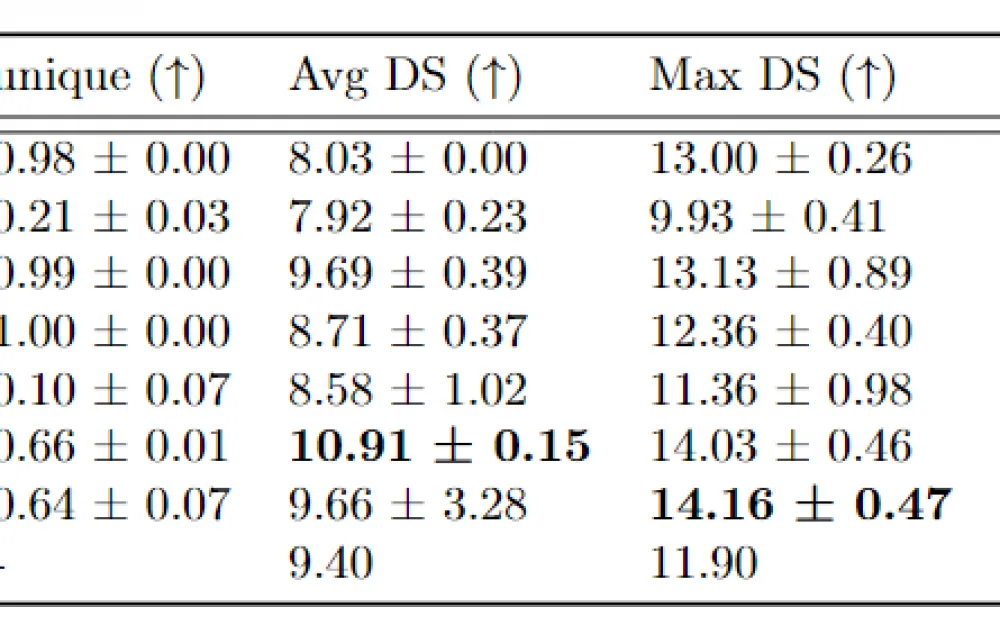
Our results show that the FFREED and FREED++ models are capable of generating molecules with higher affinity values than existing approaches.
Conclusion
Thus, it seems that we have managed to correct, simplify, and improve the FREED model quite well. However, it would be disingenuous to claim that we have reached the pinnacle of perfection in the task of generating drug molecules using deep learning methods.
As I mentioned, RL is just one of the methods. New works in the field of molecular generation are published quarterly, and today the arsenal of tools for drug discovery is quite large.
In my opinion, the most interesting at the moment are diffusion models, which restore the joint distribution of atoms of small molecules and proteins. However, they are limited by existing datasets and are also unable (without additional modifications) to optimize binding energy, which is a key property of a drug. Moreover, most of them work only with a small piece (pocket) of the protein. A more realistic approach should consider the entire protein, for example, as done in AlphaFold 3.
The second promising class of methods is generative flow neural networks. They can be considered a more efficient variant of reinforcement learning, allowing for the generation of more diverse molecules with high values of the properties of interest. However, these methods are not without drawbacks: developing a reliable reward function that reflects all the desired properties of a molecule is a complex task. The most promising direction, as it seems to me, will be the development of methods that combine these two approaches.
The implementation of FFREED and FREED++ is available on our github, we would appreciate your stars and forks!







Write comment